
آموزش ماژول BioSQL در برنامه نویسی بایوپایتون

آموزش ماژول BioSQL در برنامه نویسی بایوپایتون
در این درس از آموزش های برنامه نویسی سایت سورس باران، ما در مورد آموزش ماژول BioSQL در برنامه نویسی بایوپایتون بحث خواهیم کرد.
پیشنهاد ویژه : پکیج آموزش پایتون
BioSQL یک طرح کلی پایگاه اطلاعاتی است که عمدتا برای ذخیره توالی ها و داده های مربوط به آن برای تمام موتورهای RDBMS طراحی شده است. به گونه ای طراحی شده است که داده های همه پایگاه های اطلاعاتی بیوانفورماتیک معروف مانند GenBank ،Swissport و غیره را در خود نگه می دارد. همچنین می تواند برای ذخیره داده های داخلی نیز مورد استفاده قرار گیرد.
BioSQL در حال حاضر طرحواره خاصی را برای پایگاه داده های زیر فراهم می کند:
- MySQL (biosqldb-mysql.sql)
- PostgreSQL (biosqldb-pg.sql)
- Oracle (biosqldb-ora/*.sql)
- SQLite (biosqldb-sqlite.sql)
همچنین از پایگاه داده های HSQLDB و Derby مبتنی بر جاوا کمترین پشتیبانی را می کند.
بایوپایتون قابلیت های ORM بسیار ساده، آسان و پیشرفته را برای کار با پایگاه داده مبتنی بر BioSQL فراهم می کند. ماژول BioSQL در برنامه نویسی بایوپایتون برای انجام عملکردهای زیر فراهم می کند –
- یک پایگاه داده BioSQL ایجاد / حذف کنید
- به یک پایگاه داده BioSQL متصل شوید
- یک پایگاه داده توالی مانند GenBank ،Swisport ، نتیجه BLAST، نتیجه Entrez و غیره را تجزیه کرده و مستقیماً در پایگاه داده BioSQL بارگذاری کنید
- داده های توالی را از پایگاه داده BioSQL واکشی کنید
- داده های طبقه بندی را از NCBI BLAST واکشی کرده و در پایگاه داده BioSQL ذخیره کنید
- هرگونه جستجوی SQL را در برابر پایگاه داده BioSQL اجرا کنید
بررسی اجمالی برنامه پایگاه داده BioSQL
قبل از اینکه BioSQL را کاملا بررسی کنیم، اجازه دهید اصول برنامه BioSQL را درک کنیم. طرح BioSQL بیست و پنج جدول برای نگهداری داده های توالی، ویژگی توالی، دسته توالی / هستی شناسی و اطلاعات طبقه بندی فراهم می کند. برخی از جداول مهم به شرح زیر است:
- biodatabase
- bioentry
- biosequence
- seqfeature
- taxon
- taxon_name
- antology
- term
- dxref
ایجاد پایگاه داده BioSQL
در این بخش، بیایید با استفاده از طرحواره ارائه شده توسط تیم BioSQL، یک پایگاه داده BioSQL ایجاد کنیم. ما باید با پایگاه داده SQLite کار کنیم زیرا شروع کار بسیار آسان است و تنظیمات پیچیده ای ندارد.
در اینجا، ما با استفاده از مراحل زیر یک پایگاه داده BioSQL مبتنی بر SQLite ایجاد خواهیم کرد.
مرحله 1 – موتور پایگاه داده SQLite را دانلود کرده و آن را نصب کنید.
مرحله 2 – پروژه BioSQL را از URL GitHub دانلود کنید. https://github.com/biosql/biosql
مرحله 3 – یک کنسول را باز کنید و با استفاده از mkdir یک دایرکتوری ایجاد کنید و وارد آن شوید.
|
1 2 3 |
cd /path/to/your/biopython/sample mkdir sqlite-biosql cd sqlite-biosql |
مرحله 4 – دستور زیر را برای ایجاد یک پایگاه داده جدید SQLite اجرا کنید.
|
1 2 3 4 |
> sqlite3.exe mybiosql.db SQLite version 3.25.2 2018-09-25 19:08:10 Enter ".help" for usage hints. sqlite> |
مرحله 5 – فایل biosqldb-sqlite.sql را از پروژه BioSQL کپی کنید (/ sql / biosqldb-sqlite.sql`) و آن را در فهرست فعلی ذخیره کنید.
مرحله 6 – دستور زیر را برای ایجاد همه جداول اجرا کنید.
|
1 |
sqlite> .read biosqldb-sqlite.sql |
اکنون، تمام جداول در پایگاه داده جدید ما ایجاد شده اند.
مرحله 7 – دستور زیر را برای دیدن تمام جداول جدید در پایگاه داده خود اجرا کنید.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 |
sqlite> .headers on sqlite> .mode column sqlite> .separator ROW "\n" sqlite> SELECT name FROM sqlite_master WHERE type = 'table'; biodatabase taxon taxon_name ontology term term_synonym term_dbxref term_relationship term_relationship_term term_path bioentry bioentry_relationship bioentry_path biosequence dbxref dbxref_qualifier_value bioentry_dbxref reference bioentry_reference comment bioentry_qualifier_value seqfeature seqfeature_relationship seqfeature_path seqfeature_qualifier_value seqfeature_dbxref location location_qualifier_value sqlite> |
سه دستور اول، دستورات پیکربندی برای پیکربندی SQLite است تا نتیجه را به صورت قالب بندی شده نشان دهد.
مرحله 8 – نمونه فایل GenBank ، ls_orchid.gbk ارائه شده توسط تیم بایوپایتون https://raw.githubusercontent.com/biopython/biopython/master/Doc/examples/ls_orchid.gbk را در فهرست فعلی کپی کرده و به عنوان orchid.gbk ذخیره کنید .
مرحله 9 – با استفاده از کد زیر یک اسکریپت پایتون، load_orchid.py ایجاد کنید و آن را اجرا کنید
|
1 2 3 4 5 6 7 8 9 |
from Bio import SeqIO from BioSQL import BioSeqDatabase import os server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db") db = server.new_database("orchid") count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit() server.close() |
کد فوق رکورد موجود در فایل را تجزیه کرده و آن را به اشیا پایتون تبدیل کرده و در پایگاه داده BioSQL قرار می دهد. ما در بخش بعدی کد را تجزیه و تحلیل خواهیم کرد.
سرانجام، ما یک پایگاه داده جدید BioSQL ایجاد کردیم و برخی از داده های نمونه را در آن بارگذاری کردیم.
نمودار ER ساده
جدول biodatabase در بالای سلسله مراتب قرار دارد و هدف اصلی آن سازماندهی مجموعه ای از داده های توالی در یک گروه / پایگاه داده مجازی است. هر ورودی در پایگاه داده زیستی به یک پایگاه داده جداگانه اشاره دارد و با پایگاه داده دیگری مخلوط نمی شود. تمام جداول مربوط به پایگاه داده BioSQL به ورودی پایگاه داده زیستی اشاره دارند.
جدول bioentry تمام جزئیات مربوط به یک دنباله را به جز داده های توالی در خود دارد. داده های توالی یک biosequence خاص در جدول توالی زیست ذخیره می شود.
taxon و taxon_name جزئیات طبقه بندی هستند و هر ورودی برای مشخص کردن اطلاعات طبقه بندی خود، این جدول را ارجاع می دهد.
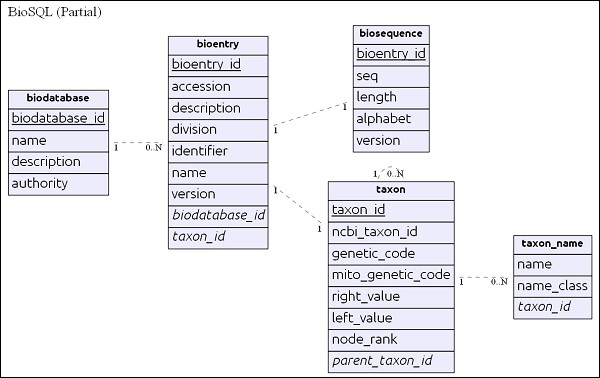
پس از درک طرح، اجازه دهید برخی از سوالات موجود در بخش بعدی را بررسی کنیم.
کوئری های BioSQL
اجازه دهید برای درک بهتر نحوه سازماندهی داده ها و ارتباط جداول با یکدیگر ، به برخی کوئری های BioSQL بپردازیم. قبل از ادامه، اجازه دهید پایگاه داده را با استفاده از دستور زیر باز کنیم و برخی از دستورات قالب بندی را تنظیم کنیم –
|
1 2 3 4 5 |
> sqlite3 orchid.db SQLite version 3.25.2 2018-09-25 19:08:10 Enter ".help" for usage hints. sqlite> .header on sqlite> .mode columns |
header. و mode. گزینه های قالب بندی برای تجسم بهتر داده ها هستند. برای اجرای درخواست می توانید از هر ادیتور SQLite نیز استفاده کنید.
به شرح زیر پایگاه داده توالی مجازی موجود در سیستم را لیست کنید:
|
1 2 3 4 5 6 7 8 9 10 11 |
select * from biodatabase; *** Result *** sqlite> .width 15 15 15 15 sqlite> select * from biodatabase; biodatabase_id name authority description --------------- --------------- --------------- --------------- 1 orchid sqlite> |
در اینجا، ما فقط یک پایگاه داده داریم، orchid.
با کد ارائه شده در زیر، ورودی های موجود در پایگاه داده orchid را لیست کنید
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 |
select be.*, bd.name from bioentry be inner join biodatabase bd on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1, 3; *** Result *** sqlite> .width 15 15 10 10 10 10 10 50 10 10 sqlite> select be.*, bd.name from bioentry be inner join biodatabase bd on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' Limit 1,3; bioentry_id biodatabase_id taxon_id name accession identifier division description version name --------------- --------------- ---------- ---------- ---------- ---------- ---------- ---------- ---------- ----------- ---------- --------- ---------- ---------- 2 1 19 Z78532 Z78532 2765657 PLN C.californicum 5.8S rRNA gene and ITS1 and ITS2 DN 1 orchid 3 1 20 Z78531 Z78531 2765656 PLN C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DN 1 orchid 4 1 21 Z78530 Z78530 2765655 PLN C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 D 1 orchid sqlite> |
جزئیات توالی مربوط به یک ورودی (accession − Z78530, name − C. fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DNA) را با کد داده شده فهرست کنید:
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 |
select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq, bs.length, be.accession, be.description, bd.name from biosequence bs inner join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532'; *** Result *** sqlite> .width 15 5 10 50 10 sqlite> select substr(cast(bs.seq as varchar), 0, 10) || '...' as seq, bs.length, be.accession, be.description, bd.name from biosequence bs inner join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532'; seq length accession description name ------------ ---------- ---------- ------------ ------------ ---------- ---------- ----------------- CGTAACAAG... 753 Z78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA orchid sqlite> |
توالی کامل مرتبط با یک ورودی (accession − Z78530, name − C. fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DNA) را با استفاده از کد زیر دریافت کنید
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 |
select bs.seq from biosequence bs inner join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532'; *** Result *** sqlite> .width 1000 sqlite> select bs.seq from biosequence bs inner join bioentry be on be.bioentry_id = bs.bioentry_id inner join biodatabase bd on bd.biodatabase_id = be.biodatabase_id where bd.name = 'orchid' and be.accession = 'Z78532'; seq ---------------------------------------------------------------------------------------- ---------------------------- CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTGAATCT GGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGGGCCTCC TCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGCATCACTGATGAATGACATTATTGT CAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAATTTTTATGACTCTCGCAACGGATATCTTGGCTC TAACATCGATGAAGAACGCAG sqlite> |
طبقه بندی های مرتبط با پایگاه داده زیستی orchid را لیست کنید
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 |
select distinct tn.name from biodatabase d inner join bioentry e on e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id = e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name = 'orchid' limit 10; *** Result *** sqlite> select distinct tn.name from biodatabase d inner join bioentry e on e.biodatabase_id = d.biodatabase_id inner join taxon t on t.taxon_id = e.taxon_id inner join taxon_name tn on tn.taxon_id = t.taxon_id where d.name = 'orchid' limit 10; name ------------------------------ Cypripedium irapeanum Cypripedium californicum Cypripedium fasciculatum Cypripedium margaritaceum Cypripedium lichiangense Cypripedium yatabeanum Cypripedium guttatum Cypripedium acaule pink lady's slipper Cypripedium formosanum sqlite> |
بارگیری داده در پایگاه داده BioSQL
بیایید بفهمیم که چگونه داده های توالی را در این بخش در پایگاه داده BioSQL بارگذاری کنیم. ما قبلاً کدی برای بارگذاری داده ها در پایگاه داده در بخش قبلی داریم و کد به شرح زیر است –
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
from Bio import SeqIO from BioSQL import BioSeqDatabase import os server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db") DBSCHEMA = "biosqldb-sqlite.sql" SQL_FILE = os.path.join(os.getcwd(), DBSCHEMA) server.load_database_sql(SQL_FILE) server.commit() db = server.new_database("orchid") count = db.load(SeqIO.parse("orchid.gbk", "gb"), True) server.commit() server.close() |
ما نگاه عمیق تری به هر خط از کد و هدف آن خواهیم داشت –
خط 1 – ماژول SeqIO را بارگیری می کند.
خط 2 – ماژول BioSeqDatabase را بارگیری می کند. این ماژول تمام قابلیت های تعامل با پایگاه داده BioSQL را فراهم می کند.
خط 3 – ماژول سیستم عامل را بارگیری می کند.
خط 5 – open_database با درایور پیکربندی شده (driver) پایگاه داده مشخص شده (db) را باز کرده و یک دسته را به پایگاه داده server) BioSQL) باز می گرداند. بایوپایتون از پایگاه داده sqlite ،mysql ،postgresql و oracle پشتیبانی می کند.
خط 6-10 – متد load_database_sql sql را از فایل خارجی بارگیری کرده و آن را اجرا می کند. ما می توانیم از این مرحله صرف نظر کنیم زیرا ما قبلاً پایگاه داده را با برنامه ایجاد کرده ایم.
خط 12 – متد های new_database پایگاه داده مجازی جدیدی را ایجاد می کند، orchid و یک دسته db را برمی گرداند تا دستور را در برابر پایگاه داده orchid اجرا کند.
خط 13 – متد load ورودی های توالی (قابل تکرار SeqRecord) را در پایگاه داده orchid بارگیری می کند. SqlIO.parse پایگاه داده GenBank را تجزیه می کند و تمام توالی های موجود در آن را به عنوان قابل تکرار SeqRecord برمی گرداند. پارامتر دوم (True) متد load به آن دستور می دهد تا جزئیات طبقه بندی داده های توالی را از وب سایت NCBI blast دریافت کند، در صورتی که از قبل در سیستم موجود نیست.
خط 14 – commit معامله را انجام می دهد.
خط 15 –closeاتصال پایگاه داده را می بندد و دسته سرور را از بین می برد.
واکشی داده های توالی
اجازه دهید دنباله ای با شناسه 2765658 را از پایگاه داده orchid به شرح زیر بیاوریم:
|
1 2 3 4 5 6 7 8 |
from BioSQL import BioSeqDatabase server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db") db = server["orchid"] seq_record = db.lookup(gi = 2765658) print(seq_record.id, seq_record.description[:50] + "...") print("Sequence length %i," % len(seq_record.seq)) |
در اینجا، سرور [“orchid”] دسته را برای واکشی داده ها از پایگاه داده مجازی orchid برمی گرداند. روش جستجوی گزینه ای برای انتخاب توالی ها بر اساس معیارها فراهم می کند و ما توالی را با شناسه 2765658 انتخاب کرده ایم. lookup اطلاعات توالی را به عنوان SeqRecordobject برمی گرداند. از آنجا که، ما قبلاً می دانیم که چگونه با SeqRecord کار کنیم ، دریافت داده ها از آن آسان است.
حذف یک پایگاه داده
حذف پایگاه داده به سادگی فراخوانی متد remove_database با نام پایگاه داده مناسب و سپس انجام آن به شرح زیر است
|
1 2 3 4 |
from BioSQL import BioSeqDatabase server = BioSeqDatabase.open_database(driver = 'sqlite3', db = "orchid.db") server.remove_database("orchids") server.commit() |
لیست جلسات قبل آموزش برنامه نویسی بایوپایتون
- آموزش برنامه نویسی بایوپایتون (Biopython)
- معرفی برنامه نویسی بایوپایتون
- آموزش نصب بایوپایتون
- ایجاد یک برنامه ساده در برنامه نویسی بایوپایتون
- آموزش دنباله در برنامه نویسی بایوپایتون
- عملیات توالی پیشرفته در برنامه نویسی بایوپایتون
- آموزش توالی ورودی/خروجی در برنامه نویسی بایوپایتون
- آموزش همترازسازی توالی در برنامه نویسی بایوپایتون
- بررسی اجمالی BLAST در برنامه نویسی بایوپایتون
- بررسی پایگاه داده Entrez در برنامه نویسی بایوپایتون
- آموزش ماژول PDB در برنامه نویسی بایوپایتون
- آموزش اشیا موتیف در برنامه نویسی بایوپایتون



.svg)
دیدگاه شما